Multiple comparisons, "Least significant difference" and Adjust P-values
Source:R/LSD.test.R
LSD.test.RdMultiple comparisons of treatments by means of LSD and a grouping of treatments. The level by alpha default is 0.05. Returns p-values adjusted using one of several methods
LSD.test( y, trt, DFerror, MSerror, alpha = 0.05, p.adj = c("none", "holm", "hommel", "hochberg", "bonferroni", "BH", "BY", "fdr"), group = TRUE, main = NULL, console = FALSE )
Arguments
| y | model(aov or lm) or answer of the experimental unit |
|---|---|
| trt | Constant( only y=model) or vector treatment applied to each experimental unit |
| DFerror | Degrees of freedom of the experimental error |
| MSerror | Means square error of the experimental |
| alpha | Level of risk for the test |
| p.adj | Method for adjusting p values (see p.adjust) |
| group | TRUE or FALSE |
| main | title of the study |
| console | logical, print output |
Value
Statistics of the model
Design parameters
Statistical summary of the study variable
Comparison between treatments
Formation of treatment groups
Details
For equal or different repetition.
For the adjustment methods, see the
function p.adjusted.
p-adj ="none" is t-student.
It is necessary first makes a analysis of variance.
if model=y, then to
apply the instruction:
LSD.test(model, "trt", alpha = 0.05,
p.adj=c("none","holm","hommel", "hochberg", "bonferroni", "BH", "BY",
"fdr"), group=TRUE, main = NULL,console=FALSE)
where the model class is
aov or lm.
References
Steel, R.; Torri,J; Dickey, D.(1997) Principles and Procedures of Statistics A Biometrical Approach. pp178.
See also
BIB.test, DAU.test,
duncan.test, durbin.test,
friedman, HSD.test, kruskal,
Median.test, PBIB.test, REGW.test,
scheffe.test, SNK.test,
waerden.test, waller.test,
plot.group
Examples
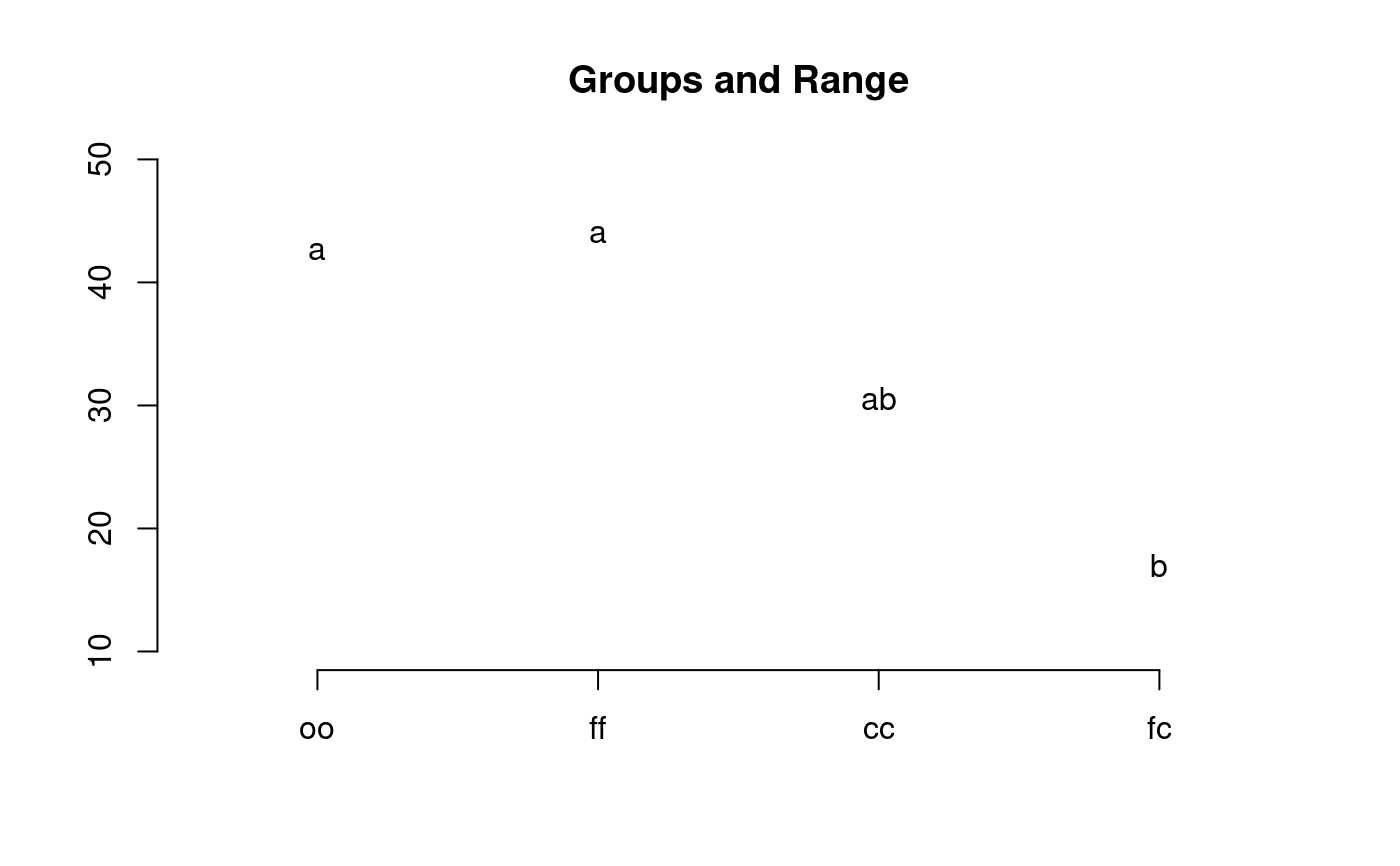
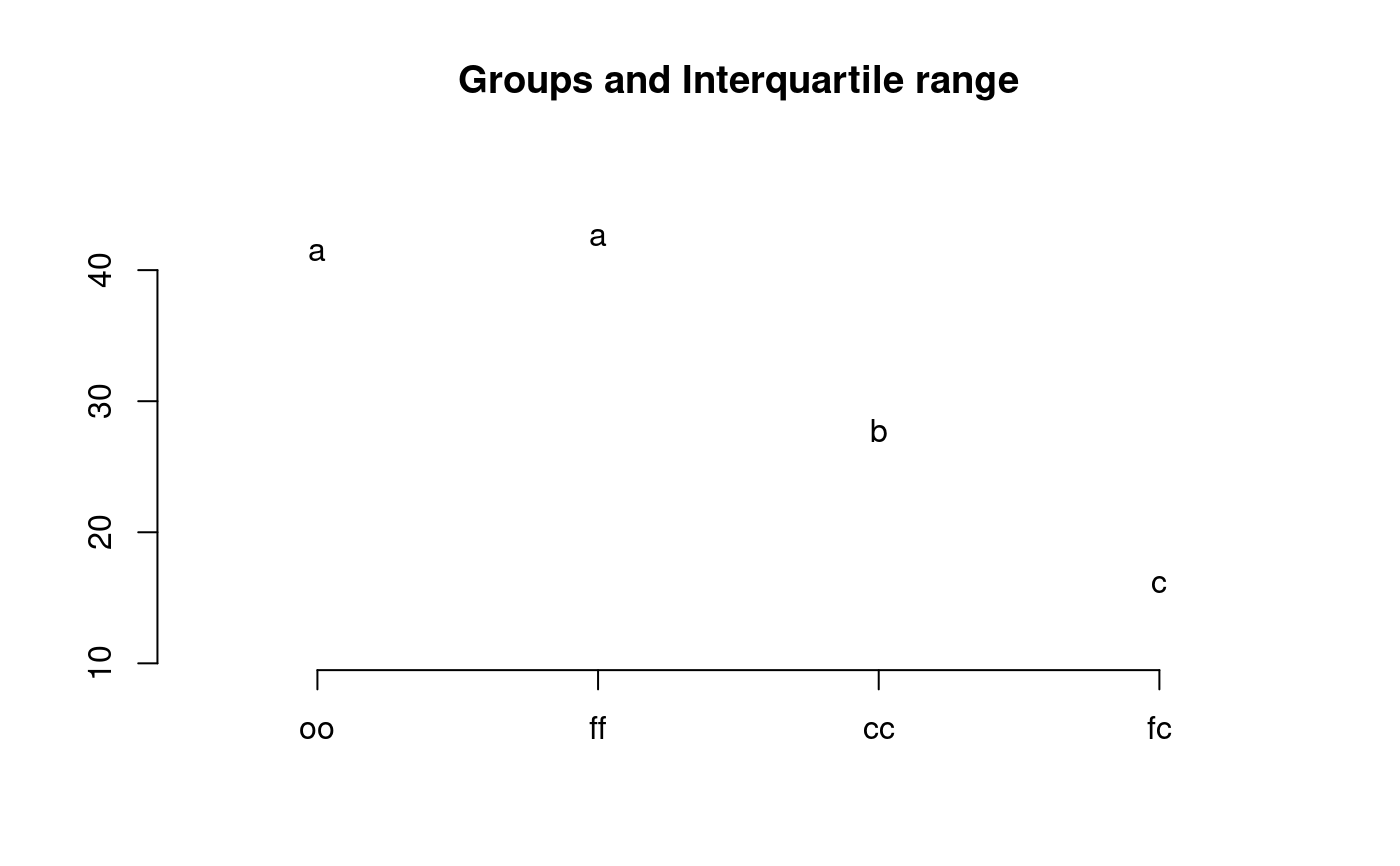
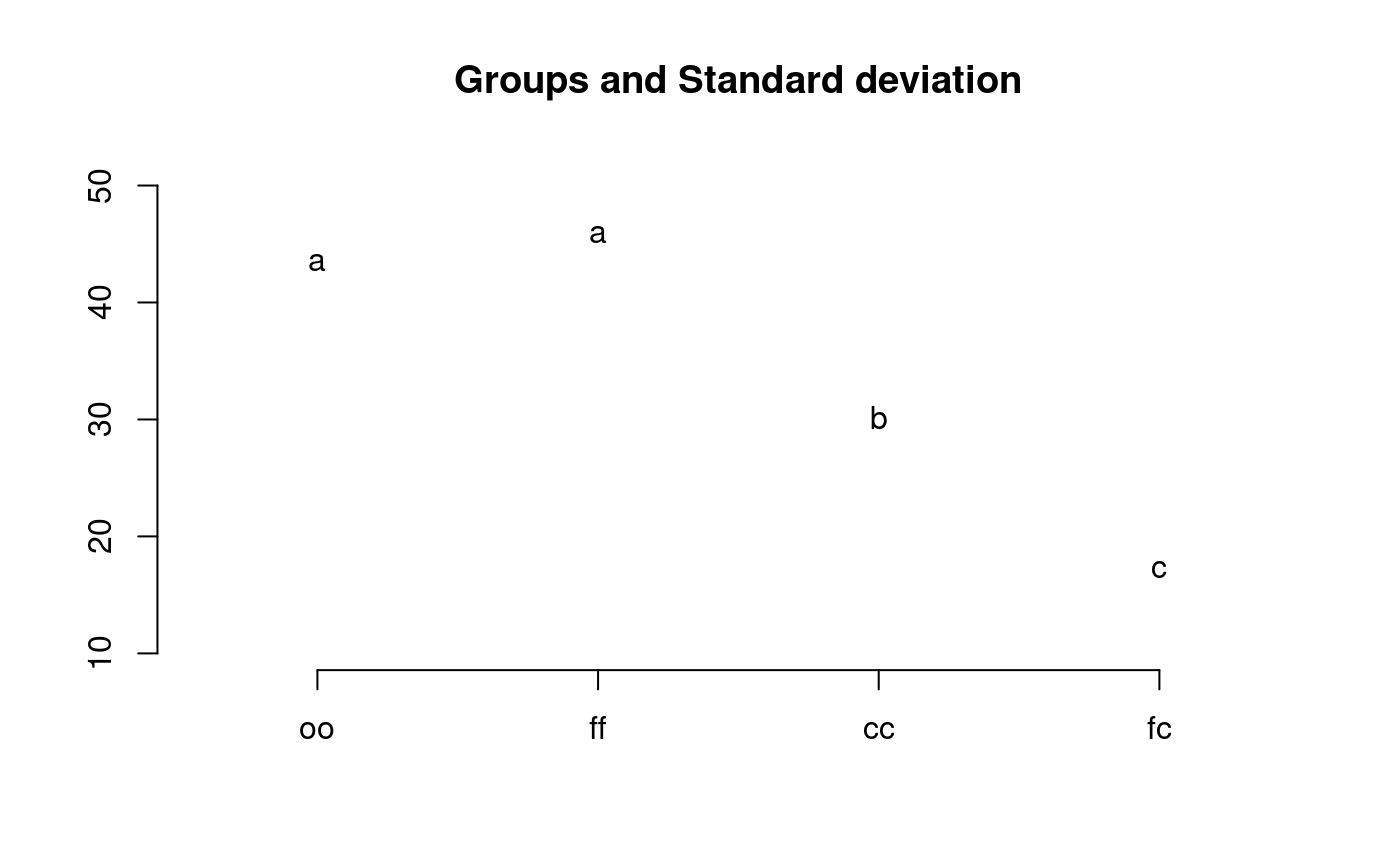
library(agricolae) data(sweetpotato) model<-aov(yield~virus, data=sweetpotato) out <- LSD.test(model,"virus", p.adj="bonferroni") #stargraph # Variation range: max and min plot(out)#> Warning: NAs introduced by coercion#endgraph # Old version LSD.test() df<-df.residual(model) MSerror<-deviance(model)/df out <- with(sweetpotato,LSD.test(yield,virus,df,MSerror)) #stargraph # Variation interquartil range: Q75 and Q25 plot(out,variation="IQR")#> Warning: NAs introduced by coercion#endgraph out<-LSD.test(model,"virus",p.adj="hommel",console=TRUE)#> #> Study: model ~ "virus" #> #> LSD t Test for yield #> P value adjustment method: hommel #> #> Mean Square Error: 22.48917 #> #> virus, means and individual ( 95 %) CI #> #> yield std r LCL UCL Min Max #> cc 24.40000 3.609709 3 18.086268 30.71373 21.7 28.5 #> fc 12.86667 2.159475 3 6.552935 19.18040 10.6 14.9 #> ff 36.33333 7.333030 3 30.019601 42.64707 28.0 41.8 #> oo 36.90000 4.300000 3 30.586268 43.21373 32.1 40.4 #> #> Alpha: 0.05 ; DF Error: 8 #> Critical Value of t: 2.306004 #> #> Minimum Significant Difference: 8.928965 #> #> Treatments with the same letter are not significantly different. #> #> yield groups #> oo 36.90000 a #> ff 36.33333 a #> cc 24.40000 b #> fc 12.86667 c#> Warning: NAs introduced by coercion